
Key Points:
- Boosting NAD+ improves memory and extends the lifespan of worms with dementia.
- Boosting NAD+ suppresses the dementia-induced alternative splicing of EVA1C, a gene necessary for memory improvements in mice with dementia.
- Alzheimer’s disease patients exhibit high levels of EVA1C, potentially due to its aberrant splicing.
To store memories of the past, the intricate structure of the brain must be delicately rearranged. Through the strengthening of some and the weakening of other neuronal connections, memories are formed, waiting to be updated by novel experience. Even into adulthood, in regions like the hippocampus, new neurons are born. This formation of new neurons—neurogenesis—is supported by a stabilizing protein called tau. When dysregulated, tau fails to maintain the tube-like structures (microtubules) necessary for neurogenesis and memory formation.
In Alzheimer’s disease (AD), dysfunctional tau proteins clump together and form tau tangles. Tau tangles are just one of the measurable risk factors associated with AD, along with aging, certain genetic mutations, inflammation, damaged mitochondria, and compromised autophagy—the process our cells use to clean up and recycle defective cellular components. Perhaps most under the radar, however, is the contribution of aberrant gene splicing to AD.
What Is Gene Splicing?
Coded within our DNA are the instructions for making every protein in the body. The journey from DNA to protein may seem simple: DNA is transcribed to a strand of messenger RNA (mRNA), which is then translated into a protein. This is called the Central Dogma of Biology. However, biology is more complicated than this simple heuristic. Indeed, biology is sufficiently complicated to stop thousands of scientists from elucidating the etiology of AD or how to effectively slow its progression. Discovering the answers to these questions may require the dismissal of scientific dogma altogether.

Tangents aside, each of our genes can be made into multiple proteins through a process called alternative splicing. For this, DNA is directly transcribed into a strand of RNA called pre-mRNA, which contains portions of genetic code called introns and exons. Introns are usually removed from pre-mRNA before creating a protein, and exons are usually retained. However, with alternative splicing, introns can be retained, and exons can be removed. Ultimately, it is the pattern of introns and exons from a single gene that determines which protein isoform is made.
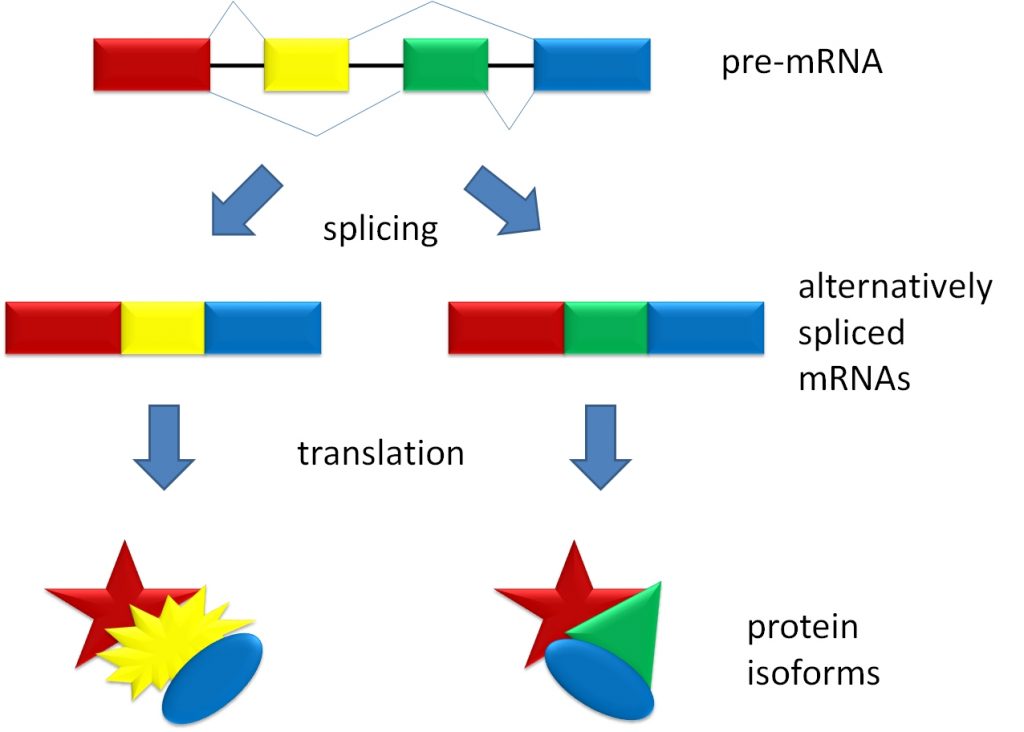
In a 2008 study, MIT researchers found that alternative splicing affects about 92-94% of human genes, suggesting nearly all of our genes can be made into multiple protein isoforms. Importantly, the function of each isoform can be related to, distinct from, or even the opposite of the other isoforms. With this in mind, and considering that properly functioning proteins are necessary for cellular health and survival, mistakes in alternative splicing can be catastrophic. Notably, alterations in the splicing machinery have been observed in AD patients, along with aberrant pre-mRNA splicing events, suggesting that aberrant alternative splicing contributes to AD pathology.
The Role of NAD+
NAD+ is a small molecule essential for mediating the production of cellular energy. Additionally, enzymes involved in DNA repair, mitochondrial health, and stress-protection, called sirtuins, use NAD+ as fuel. NAD+ levels are thought to decline under certain conditions, such as with aging, obesity, and dementia. However, since NAD+ itself is unstable, restoring NAD+ levels via oral administration requires the intake of its precursors, such as nicotinamide, NR (nicotinamide riboside), or NMN (nicotinamide mononucleotide). Both NR and NMN have been shown to improve memory in animal models of dementia. However, too few studies have explored the effects of boosting NAD+ on aberrant gene splicing in these models.
Boosting NAD+ Extends Life
In a new study to explore the effects of altered gene splicing in dementia, researchers from the University of Oslo in Norway employed mice harboring a gene from a very familiar species: humans. Rarely, humans will inherit a mutation in their tau gene called P301S, which causes a form of early-onset dementia (frontotemporal dementia) that physicians sometimes confuse with AD. In 2007, researchers from the University of Pennsylvania genetically altered mice to harbor this mutation, naming them hTau.P301S (humanized Tau.P301S) mice.
By examining the hippocampus, where memories are consolidated, of hTau.P301S mice, the Oslo researchers observed altered splicing events in genes related to mitochondrial health and neurogenesis, suggesting the dysregulation of these processes. Moving forward, to determine if boosting NAD+ reaps therapeutic effects, the researchers administered NR to the hTau.P301S mice. This led to the suppression of 20 altered splicing events, including one in a gene involved in neurogenesis called Eva1c.
Due to its potential role in memory formation, the researchers further examined Eva1c by employing worms (C. elegans). That is, worms harboring a human tau gene with a mutation very similar to P301S called P301L. Remarkably, the researchers found they could improve the memory of hTau.P301L worms by treating them with NR. What’s more, NR increased the lifespan of the hTau.P301L worms by about 17%. Interestingly, the memory enhancement and lifespan-extending effects of NR on the hTau.P301L worms disappeared when Eva1c was genetically removed.
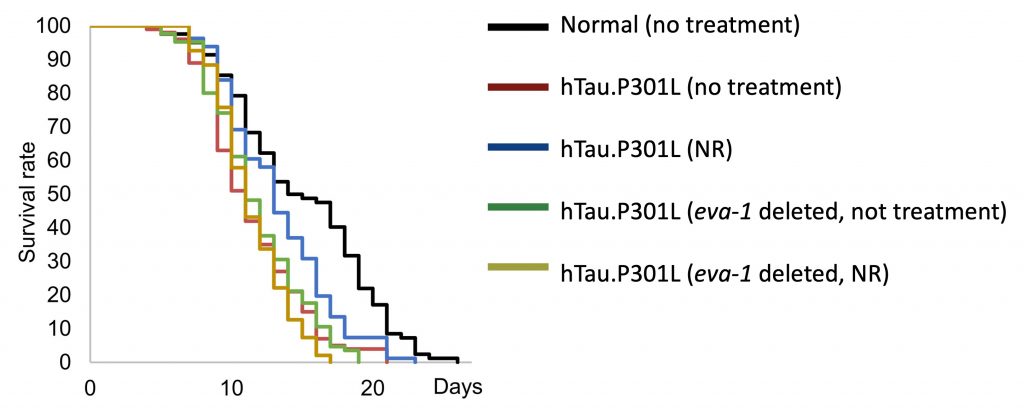
These findings suggest that the aberrant splicing of Eva1c (eva-1 in worms) may lead to memory impairments and shortened lifespan. Moreover, boosting NAD+ may potentially correct the splicing of Eva1c to improve memory and extend lifespan.
Boosting NAD+ Improves Memory
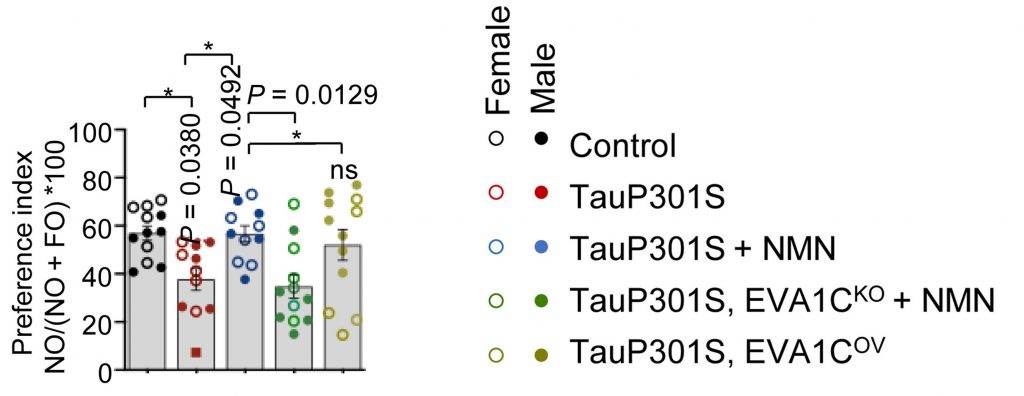
Worms have shorter lifespans and are easier to genetically manipulate than mice. However, since mice are more similar to humans, the Oslo researchers tested how Eva1c and boosting NAD+ interact to influence memory. To do so, they injected a mutated tau gene, TauP301S, into the hippocampus of normal mice. As expected, this led to memory impairments. However, treating these mice with NMN prevented the memory impairments.
Furthermore, to determine the role of EVA1C, the human version of mouse Eva1c and worm eva-1, the researchers increased its activation in the hippocampus of the mice. Strikingly, the overactivation of EVA1C, like NMN, prevented the memory impairments induced by TauP301S. Moreover, reducing EVA1C levels abolished the memory-enhancing effects of NMN. Together, these findings reveal that NMN may improve memory by correcting the splicing of EVA1C.
Human Data
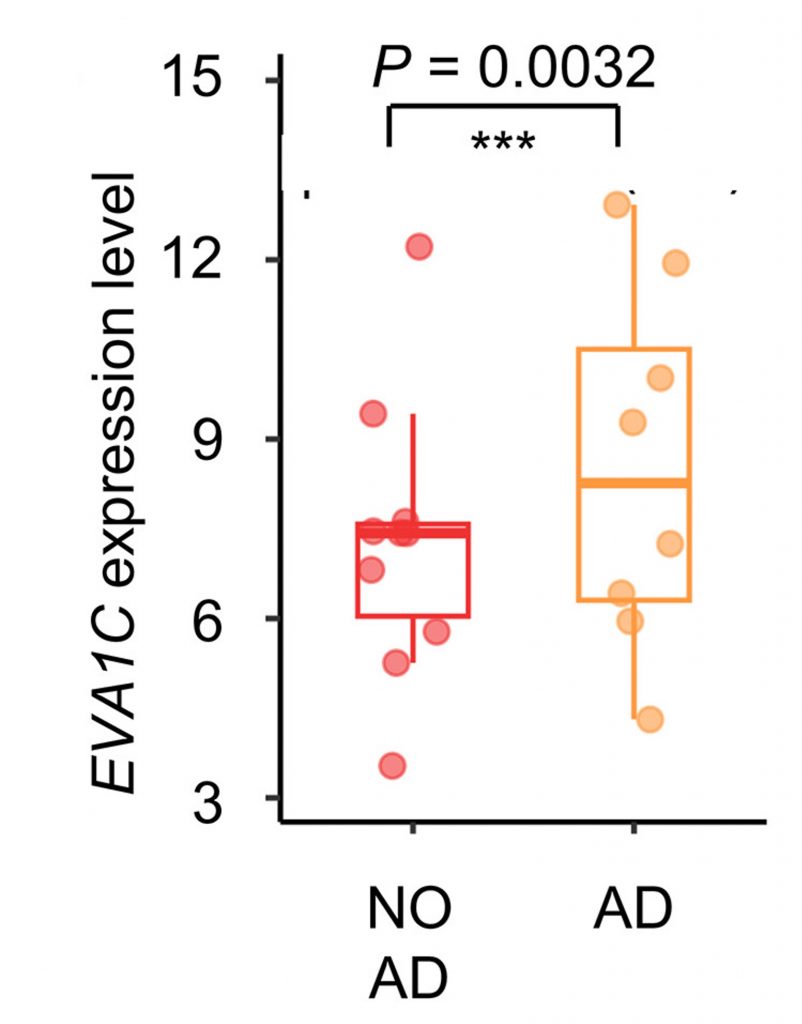
To determine the human relevance of EVA1C in dementia, the researchers examined postmortem brain tissue from AD patients. They found that EVA1C mRNA levels were higher in the brains of AD patients than individuals without AD. Together with the mouse and worm experiments, these findings support the idea that low EVA1C levels contribute to the pathology of dementia and memory loss, potentially due to aberrant splicing. In turn, boosting NAD+ levels may elevate EVA1C1 levels by correcting its splicing, leading to improved memory. In AD patients, EVA1C may be elevated as a compensatory mechanism. Nevertheless, this is only one of the first studies exploring the effects of NAD+ on alternative gene splicing, and further studies are needed to validate this line of thinking.
Halting Age-Related Memory Loss with NAD+
Many animal studies have shown that boosting NAD+ can improve memory. Additionally, a previous study showed that combining 1 g of NR with other “metabolic activators” (2.55 g of N-acetyl-L-cysteine, 3.73 g of L-carnitine tartrate, and 12.35 g of L-serine) improves memory and reverses neurodegeneration in AD patients. However, 1 g of NR alone did significantly improve the memory of individuals with mild cognitive impairment or peripheral artery disease. This could mean that combining NAD+ with other compounds is necessary to improve memory, or that cognitive deficits must be sufficiently severe to see the benefits of boosting NAD+.
 Comments
Comments