
Mitochondria are unique structures present in all cells that are essential to producing energy and stimulating growth. Malfunctional mitochondria can promote the development of degenerative brain conditions, such as Parkinson’s disease and Leigh syndrome, a dreadful neurological disorder that leads to progressive decreases in mental and movement capabilities that typically result in childhood mortalities. Unraveling the various roles of mitochondria for the preservation of brain health remains an obstacle.
In a study published in Cell Metabolism, researchers at Northwestern University Feinberg School of Medicine sought to identify the various mitochondrial functions necessary for optimal brain health by modeling Leigh syndrome with genetically altered mice that did not contain a critical brain mitochondrial protein complex. They discovered that mice had improved survival upon the introduction of a yeast protein incapable of generating energy but similar to the critical mouse mitochondrial protein complex in the brains of mice. However, this treatment failed to combat onset movement and balance problems (i.e., ataxia). The obtained results indicate that stimulation of this critical mitochondrial protein complex in the brain promotes organismal survival through its various functions rather than energy production.
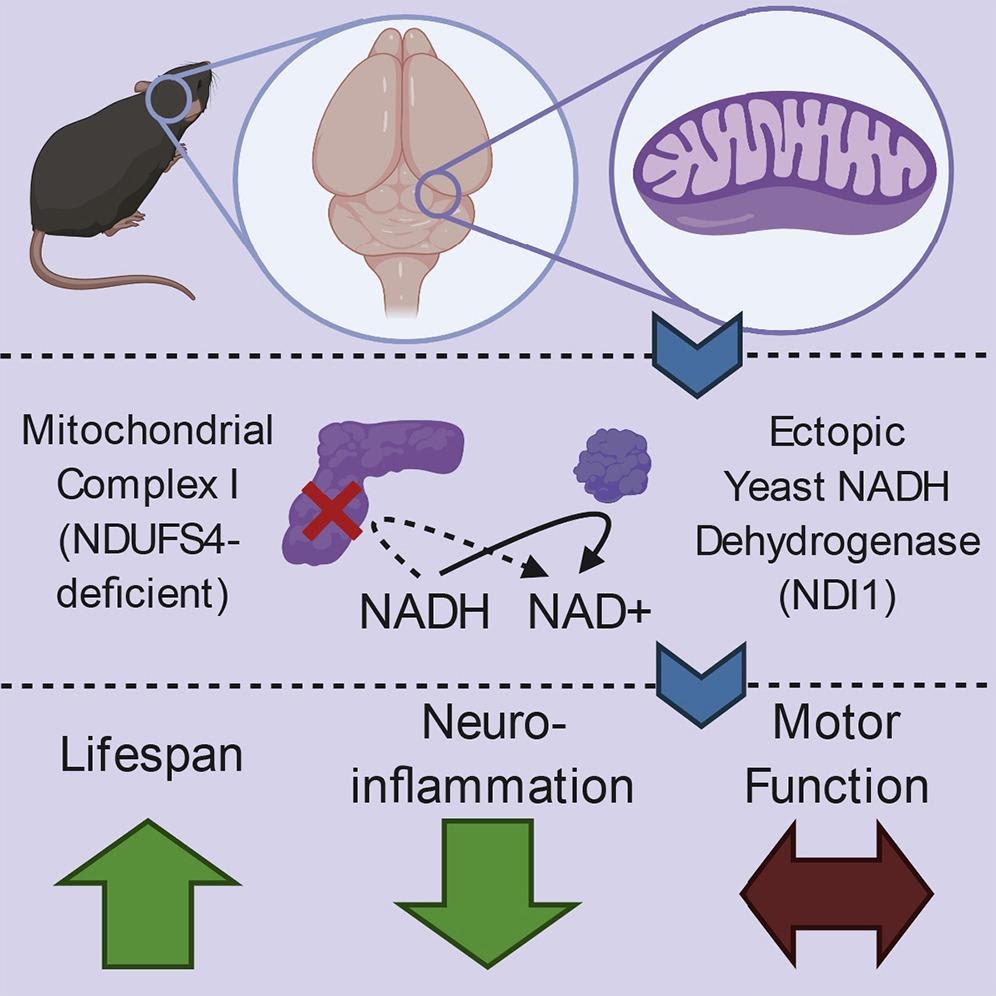
Nicotinamide adenine dinucleotide (NAD+), a critical coenzyme that mediates a variety of metabolic pathways via the transfer of electrons between its two forms NAD+ and NADH, is required for the generation of mitochondrial energy and the production of cellular building blocks. NAD+ levels are restored following the transfer of electrons from NADH molecules into the mitochondria and electron transport chain, a cluster of proteins that promotes energy production through the shuttling of electrons via an inner mitochondrial membrane. On top of promoting cellular energy production, the change of NADH to NAD+ through the mitochondrial electron transport chain facilitates the generation of building blocks for lipids, carbohydrates, amino acids, and nucleic acids.
Mitochondrial complex 1 (MC1), a collection of proteins that configures into a single multimolecular machine, is an essential factor of these processes, contributing to the restoration of NAD+ and energy production. In addition, neurodegenerative disorders such as Parkinson’s disease and Leigh syndrome have been linked to malfunctional MC1. Given that MC1 is critical for both energy production and restoration of NAD+, determining which of these tasks governs the effects of MC1 malfunction in complex tissues such as the brain remains a challenge. For these reasons, it is still unclear whether energy production or restoration of NAD+ relies on MC1 stimulation for brain function.
The investigators created a mouse model of Leigh syndrome exhibiting the absence of a pivotal subunit of MC1 in the brain that instead generated a comparable protein from yeast with limited functionality in order to figure out which of the mitochondria’s various functions is necessary for brain health in mice. Furthermore, they utilized the yeast protein NADH dehydrogenase (NDI1), a unique enzyme capable of restoring MC1’s ability to regenerate NAD+ but does not facilitate energy production. “We sought to determine whether NAD+ regeneration or bioenergetics is the dominant function of MC1 in protection from brain pathology,” said the authors.
The investigators showed that the yeast NDI1 protein, which restores NAD+ without facilitating energy production, is adequate to prolong survival but does not combat ataxia induced by brain MC1 dysfunction. Furthermore, the Leigh syndrome model demonstrated that the presence of NDI1 in the absence of MC1 was adequate to significantly extend longevity without enhancing motor function.
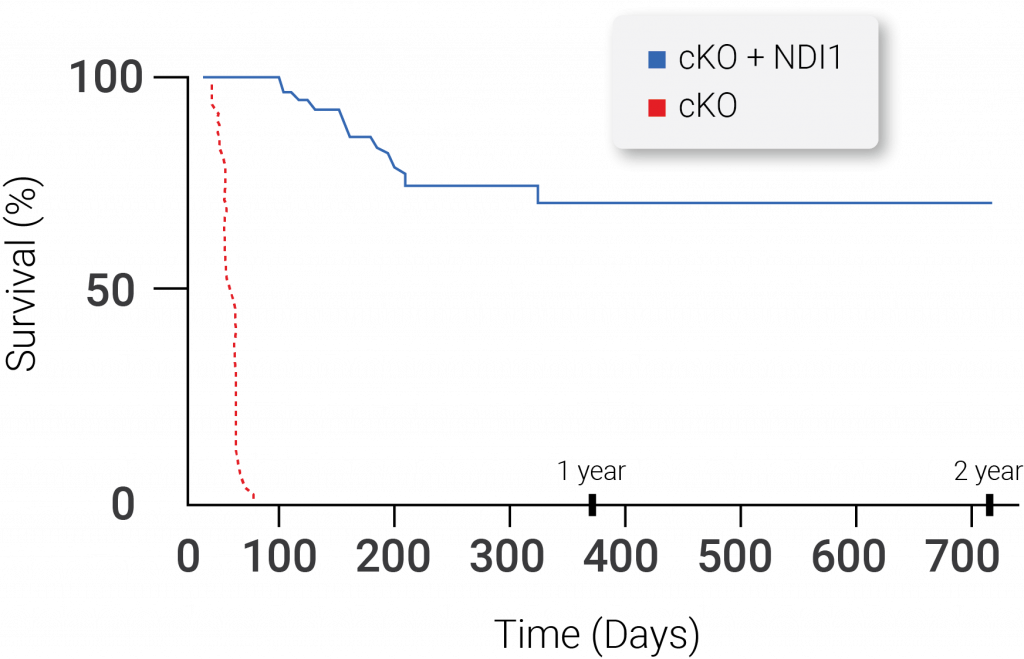
The Yeast NDI1 Protein Is Sufficient to Rescue Lifespan in a Mouse Model of Leigh Syndrome Due to Loss of Mitochondrial Complex I Function. Mice with Leigh syndrome live between 45-60 days, but, when yeast NDI1 protein is introduced, the median survival of these mice is greater than one year. The dotted line represents mice with Leigh Syndrome without mitochondrial complex I (MC1) function in the brain. The solid line represents mice without MC1 function in the brain that had the yeast NDI1 protein introduced (McElroy et al., 2020 | Cell Metabolism).
Accordingly, MC1 activity in the brain promotes survival through its ability to restore NAD+, while MC1’s bioenergetics function is needed for favorable motor control. “Overall, our results indicate that a single yeast enzyme capable of regenerating mitochondrial NAD+ from NADH is sufficient to increase the lifespan, but not maintain the motor function, of mice with impairment of MC1 in the brain,” said the authors.
 Comments
Comments