
Key Points:
- Deleting CD38 from mice restores heart function by correcting calcium homeostasis.
- Respiratory and muscle function are improved by deleting CD38.
- Inhibiting CD38 in human muscle cells corrects calcium homeostasis.
Like buttress reinforcing the walls of a cathedral, there are proteins that reinforce the structure of our cells. When these proteins are compromised, cells begin to collapse, inevitability leading to organ dysfunction. Duchenne muscular dystrophy (DMD) is caused by a compromised dystrophin protein, leading to the structural collapse of muscle cells with eventual heart and muscle impairments. DMD is currently incurable, but a recent study reveals that inhibiting an immune enzyme called CD38 could improve DMD heart and muscle function.
Published in EMBO Molecular Medicine, Zelicourt and colleagues from the University of Paris-Saclay in France report that eliminating the function of CD38 increases NAD+ levels and improves heart and muscle function in mouse models for DMD. Heart function is fully restored by correcting for deficits in calcium homeostasis. Calcium homeostasis is also corrected in human muscle cells from DMD patients by inhibiting CD38 with the drug isatuximab. While the correct CD38 inhibitor still needs to be determined, CD38 inhibition may be a promising therapy for DMD.
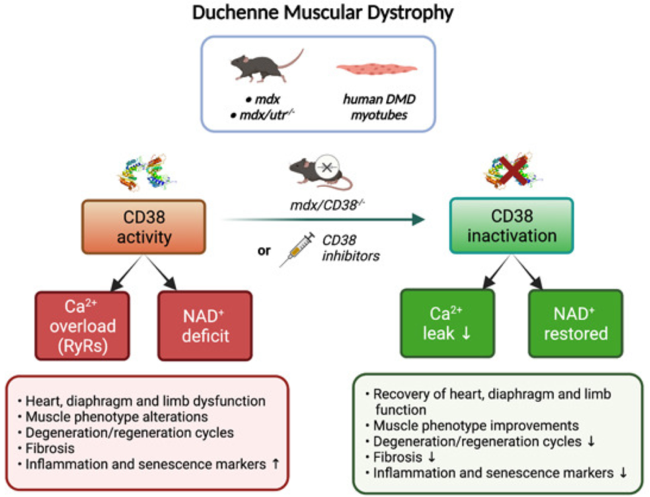
(Zelicourt et al., 2022 | EMBO Molecular Medicine) Eliminating CD38 Corrects Duchenne Muscular Dystrophy (DMD) Pathology. In two mouse models for DMD (mdx and mdx/utr-/-) and human DMD muscle cells (myotubes), CD38 depletes NAD+ and dysregulates calcium homeostasis. Genetic deletion or drug inhibition of CD38 restores NAD+ and calcium homeostasis leading to improvements in heart and muscle function.
DMD Skeletal and Cardiac Muscle Degeneration
DMD is a rare but devastating disease. Considerable care is needed just to push a patient’s life expectancy to the age of 30. Due to a compromised dystrophin protein, DMD muscle cells’ structural integrity are weakened, making the cells more prone to damage. Over the course of the disease, these fragile muscle cells progressively die off, leading to muscle degeneration. This includes skeletal and cardiac muscle degeneration, both of which were examined in this study.
Skeletal muscle is the type of muscle that we usually think of when we hear the term “muscle” and is the muscle we have voluntary control over. DMD patients are often restricted to a wheelchair because their lower limb muscles are too weak to allow for walking. DMD patients also have respiratory problems because of their weakened diaphragm, the muscle that controls our breathing. Another type of muscle, called cardiac muscle, is the muscle that contracts in our heart to make it beat. Though all muscles are severely altered in DMD, cardiac muscle dysfunction and heart failure are the main causes of DMD patient death.
The DMD NAD+ Connection: CD38
Recently, NAD+ – a vital molecule responsible for cell energy homeostasis – was shown to be reduced in DMD, leading to muscle dysfunction. However, the connection between NAD+ and DMD muscle pathology has remained unclear. Calcium (Ca2+) – the signaling molecule that mediates the contraction of muscle – is dysregulated in DMD muscle cells, central to the development of muscle weakness, cardiac dysfunction, and muscle cell death. Thus, CD38 – an enzyme that breaks down NAD+ and modulates calcium signaling – connects NAD+ to DMD. CD38 is part of the immune defense system but can have detrimental effects when chronically activated, such as with aging. Activation of CD38 may be the primary driver of NAD+ decline during aging. While CD38 is the major NAD+ consumer in the heart and muscle, it has yet to be explored in the context of DMD.
CD38 Deletion Improves Skeletal Muscle Function
To investigate the role of CD38 in DMD, Zelicourt and colleagues genetically deleted the CD38 gene from the most commonly used mouse model for DMD, called the mdx mouse. The researchers evaluated NAD+ levels from the cardiac and skeletal muscle of old mdx mice. The heart, diaphragm, and lower limb muscles of mdx mice had dramatically reduced levels of NAD+. This was fully restored by deletion of CD38, demonstrating that CD38 is the primary mediator of NAD+ depletion in DMD cardiac and skeletal muscle.
DMD patients older than 18 suffer from heart dysfunction. Similarly, mdx mice show a progressive development of heart defects from 6 months of age. Zelicourt and colleagues found that deleting CD38 offered full protection against heart dysfunction, demonstrating that boosting NAD+ by eliminating CD38 could prevent DMD heart pathology.
Calcium homeostasis – the normal movement of calcium within, across, and outside of cells – is vital in maintaining muscle function. Zelicourt and colleagues found that, in mdx cardiac muscle cells, measures of calcium homeostasis were dysregulated. These measures included an increase in propagating waves of calcium called calcium waves. Deleting CD38 fully restored the calcium waves and other measures of calcium homeostasis to normal levels. These findings suggest that mdx heart function can be restored through the restoration of calcium homeostasis by eliminating CD38.
Respiratory function, which is dependent on the diaphragm, was found to be partially restored by deleting CD38. Lower limb muscle function, as measured by resistance to fatigue (grip duration) and strength (maximum force) was also improved by deletion of CD38. These findings support the protective effect of CD38 deletion in mdx skeletal muscle.
CD38 Inhibitors Improve DMD Pathology in Mice
As genetically deleting CD38 from mdx mice was shown to improve lower limb muscle function, Zelicourt and colleagues next tested whether pharmacological inhibition of CD38 could achieve the same effects. They treated mdx mice with a CD38 inhibitor derived from flavonoids (class of compounds in plants) called K-rhein. K-rhein restored fatigue and strength deficits in the mdx mice, similar to the effect of CD38 deletion. Histological liver and kidney features showed no signs of toxicity, suggesting the potential of K-rhein to be used as a therapy for DMD muscle function.
Another CD38 inhibitor called 78c was also used on mdx mice. The mdx mice treated with 78c showed higher skeletal muscle NAD+ levels. They also had improved resistance to fatigue, exercise performance, and respiratory function. Overall, the K-rhein and 78c CD38 inhibitor data demonstrate that many of the skeletal muscle improvements observed with CD38 deletion can be achieved with a CD38 inhibitor.
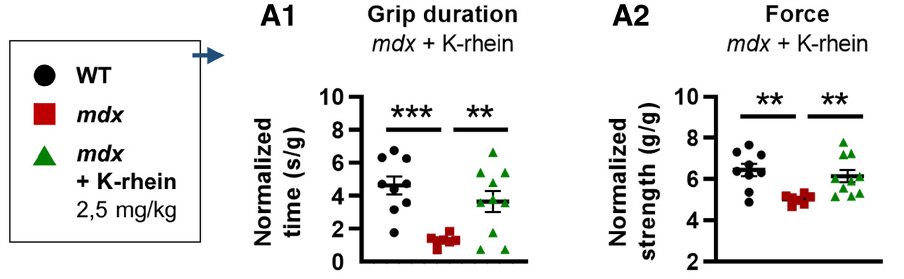
(Zelicourt et al., 2022 | EMBO Molecular Medicine) Inhibiting CD38 Improves Skeletal Muscle Function in Mouse Model for Muscular Dystrophy. (A1) Grip duration and (A2) strength are improved in mdx mouse model for muscular dystrophy (mdx) when treated with CD38 inhibitor K-rhein.
CD38 Inhibitor Corrects Calcium Homeostasis in Human DMD Cells
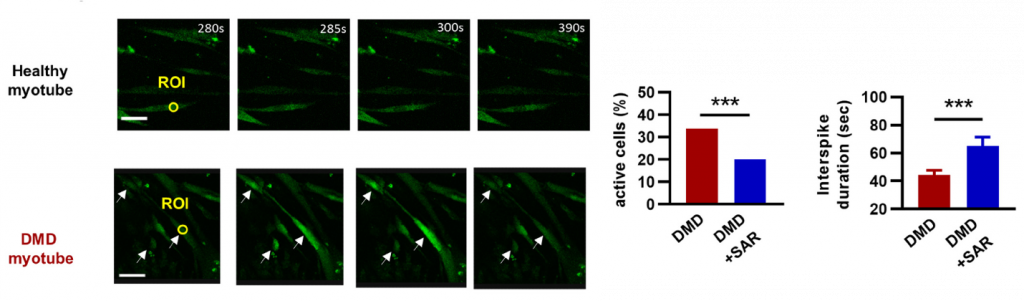
To help validate the efficacy of their results from mice, Zelicourt and colleagues treated human DMD skeletal muscle cells (myotubes) with an FDA-approved CD38 inhibitor called isatuximab. Isatuximab reduced the number of muscle cells displaying calcium waves and, in the remaining cells, reduced the frequency of waves, demonstrating a correction of calcium homeostasis. However, isatuximab failed to increase NAD+ levels.
Finding the Right CD38 Inhibitor for DMD
The therapies that are currently available for DMD have limited effects on disease onset. These therapies are very costly for individuals and caregivers. A drug that directly targets the NAD+ breakdown activity of CD38 with limited cytotoxic effects would be ideal for testing new DMD treatments. The findings of Zelicourt and colleagues demonstrate that deleting CD38 can raise NAD+ levels and improve heart and muscle function in mdx mice, but inhibiting CD38 with isatuximab in human DMD cells does not raise NAD+ levels. This is likely due to the way the drug works, as isatuximab doesn’t directly block the NAD+ breakdown activity of CD38, it is an antibody that marks cells that helm CD38 for destruction, which can potentially be harmful to muscle cells.
Thus, it seems other CD38 inhibitors need to be explored for the treatment of DMD. For example, 78c binds directly to the NAD+ breakdown site of CD38 (competitive inhibitor) and K-rhein inhibits the breakdown site by binding to CD38 elsewhere (noncompetitive inhibitor). Both 78c and K-rhein may be better candidates for DMD therapy because they inhibit CD38 directly without killing cells.
 Comments
Comments